Genomic variation: Genotyping and sequencing
Motivation
The phenotypic and genotypic characterization of plant genetic resources is the foundation for their further utilization. For example, if a breeder wants to identify a landrace of barley that is resistant to certain pathogen strain, genetic resources should be characterized for this trait to identify the landrace. In combination with large-scale genotypic characterization, the gene(s) controlling the resistance may be identified and utilized in breeding using, for example, marker assisted introgression.
This is just one example for the high value of genotypic characterization - there are many other applications for genotyping. In the following chapter, the different types of genetic variation and methods for characterization will be introduced. The focus is not on technical details, but on a broad overview. A result of the genotyping or sequencing of genetically diverse material are data, which need to be structured in a certain manner. Currently the VCF file format is state of the art in the structuring of genotyping and in particular sequencing data. Since we will be working with this format, it will be introduced in this chapter.
Learning goals
- Unterstand the types of genetic variation segregating in plant genomes
- Being able to differentiate between the information content and utility of genetic markers and genome sequences
- Understand the principle of shotgun whole genome sequencing
- Know the different types of variants and how they are represented in the VCF file format
Types of genetic variation
Genetic Polymorphisms
In plant breeding, one is mainly interested in utilizing heritable phenotypic variation. This variation results from genetic polymorphism at genes that control traits of interest.
A genetic polymorphism is defined as variation of the genotype that occurs in a population and is determined only by genetic factors (as opposed to epigenetic or environmental factors).
One of the goals of plant genetic resource management is to measure the extent of genetic polymorphism in a breeding or wild population for later utilization in plant breeding.
The study of genetic variation in the early 20th century began on the level of individuals by looking at their phenotype and phenotypic variation between individuals. As a consequence of technical progress in molecular biology, the ability to actually investigate genotypic instead of phenotypic variation became possible. The following list outlines the types of genetic polymorphisms that were used in the last century to investigate inheritance and to use the information for breeding:
- 1900’s: Visible polymorphisms
- 1930’s: Chromosomal polymorphisms
- 1940’s: Blood groups
- 1960’s: Protein polymorphisms
- 1980’s: DNA Sequencing
- 2000’s: Resequencing of genomes
Since most polymorphism types are of genetic interest only, we will focus only on the two types of polymorphisms that are used today: SNP genotyping and DNA sequencing.
In this module, we will not teach the following components of DNA and assume that you have a basic understanding of these concepts:
- Structure of DNA
- Functional elements of DNA (Genes, promotors, etc.)
- Genetic code, etc.
- Diversity of genetic markers
Although the analysis of genetic variation in form of genetic markers has a long history and contributed much to an understanding of genetic variation in natural and breeding process, in the following we will only discuss DNA sequence variation.
DNA sequence variation
Variation in the DNA is the ultimate heritable genetic variation, e.g. variation of the genotype.
Although DNA is a relatively simple, linear molecule consisting of four ‘letters’, there are several types of sequence polymorphism:
- Single nucleotide polymorphisms (SNPs)
- Insertion or Deletion variants (Indels)
- Structural genomic variants: Insertions or deletions from larger DNA seqments.
- Variation in other repetitive elements such as minisatellites or gene families.
DNA sequence variants can be detected by a variety of experimental methods. In this case, they are called markers.
An important difference is whether a marker is dominant or co-dominant. Dominant markers are not able to discriminate between homozygous and heterozygous individuals, whereas co-dominant markers allow to differentiate between heterozygous and homozygous genotypes.
The most widely used markers are SNPs, because several methods were developed that allows to determine their allelic state with high precision.
Methods for the analysis of genetic variation
SNP genotyping
In most plant breeding programs single nucleotide polymorphism (SNP) genotyping is (still) the method of choice. It consists of several steps:
- Identification of SNPs by sequencing a small panel of individuals
- Design of a SNP array using a computer program
- Genotyping of many other individuals using the SNP array
There are a number of technologies available. One key aspect of the SNP array is that usually the number of markers is investigated, ranging from 384 to about 600,000, depending on the type and size of the chip. These SNPs are genetic markers, because usually their position in the genome is known and can therefore be used to investigated genetic relationships, but also to use them for genetic mapping. A key advantage of SNP arrays is that SNPs are selected such they give a result in most individuals tested, in other words, the proportion of missing data is small.
There are numerous technologies available for the genotyping of DNA. One assay technology is shown in Figure 1. An overview of currently available methods is given by Kim and Misra (2007).
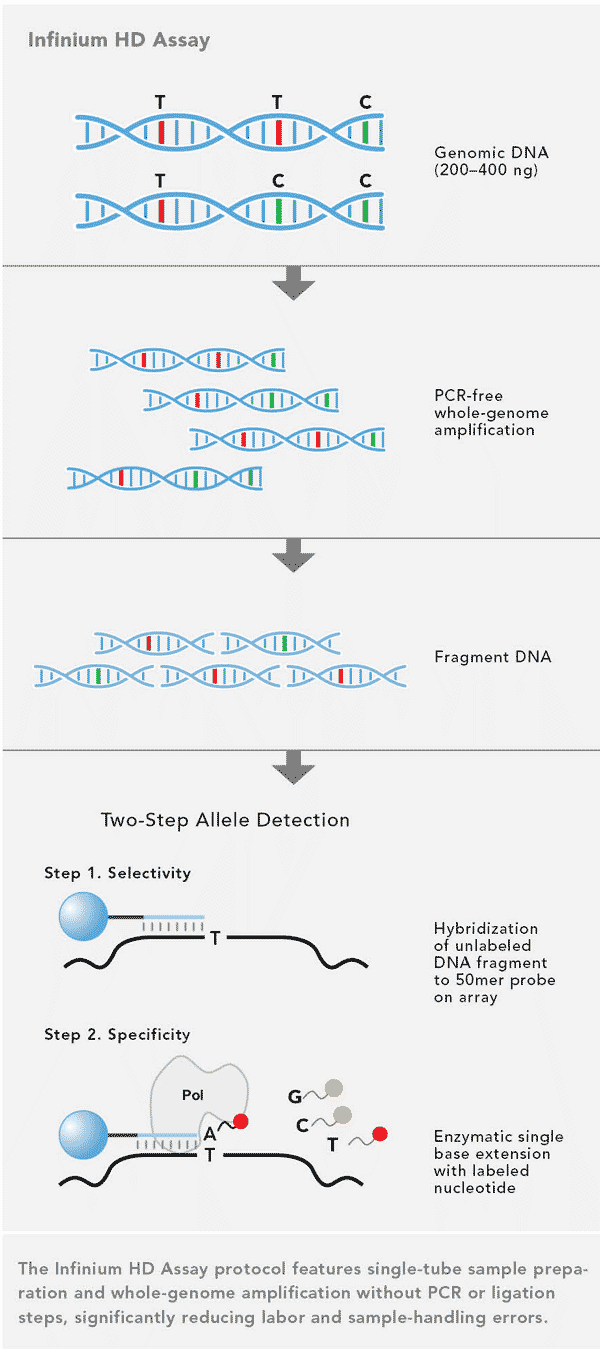
For example, the identification of SNP markers for genotyping in rye is described by Haseneyer et al. (2011). One of the largest applications of this technology is the genotyping of the complete USDA soybean collection (ca. 20,000 accessions), which was genotyped with 50.000 SNPs (Song et al., 2015). This analysis identified remarkable differences in the genetic diversity of landraces as opposed to modern elite varieties.
DNA sequencing technologies
The sequencing of DNA is the best method to characterize genetic variation segregating in a genomic region because all polymorphisms of a genome can be detected.
The classical method for DNA sequencing was developed by Frederic Sanger and is called Sanger sequencing. Since it is not widely used anymore and only for specific purposes, you are referred to the Wikipedia page.
After sequencing a certain gene or a genomic region of several individuals, a sequence alignment is produced with software programs and visualized with sequence alignment editors (Figure 2). In its simplest definition, a sequence alignment is a matrix of DNA sequences that are aligned by their homologous positions in the genome. Homology is defined as the regions (or nucleotides) that have the same evolutionary history, e.g., can be traced back to the same ancestral genomic region or sequence.
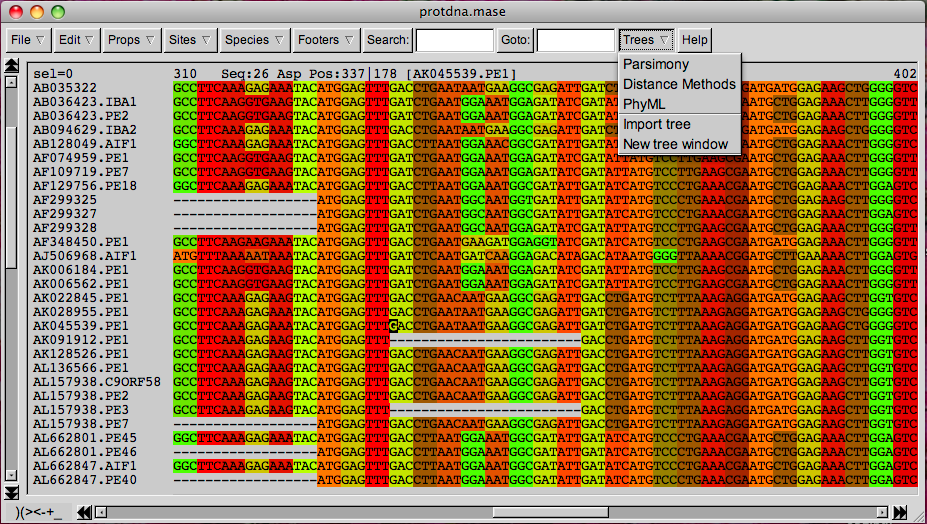
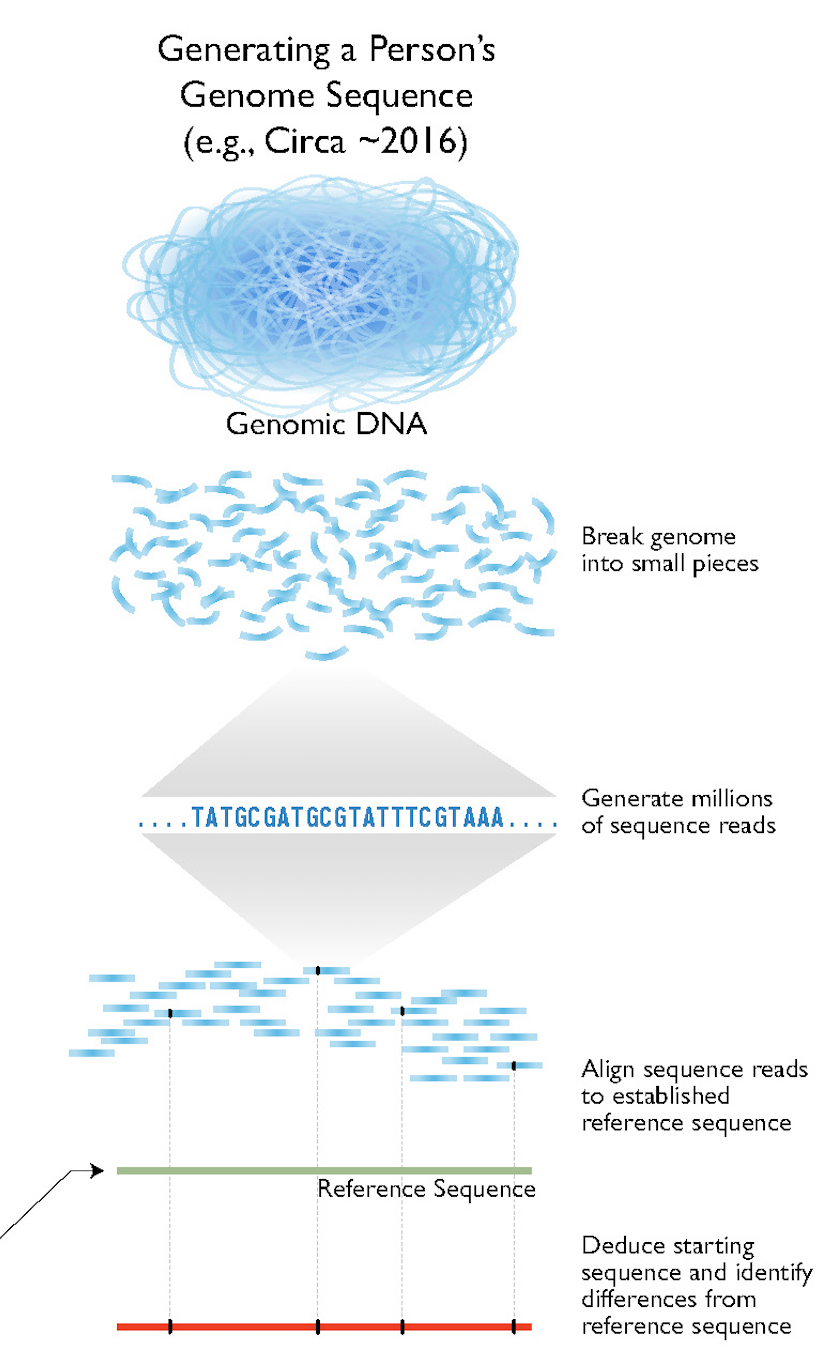
Today, the state of the art is whole genome resequencing (WGRS) of crops (Figure 6).
The sequencing of DNA can be considered to be the gold standard of the analysis of genetic variation because it has the potential to uncover the complete genetic variation present in a genome. Due to its importance, there are several technological revolutions going on that have the consequence that the price of sequencing (per base pair) is decreasing rapidly (Figure 3).
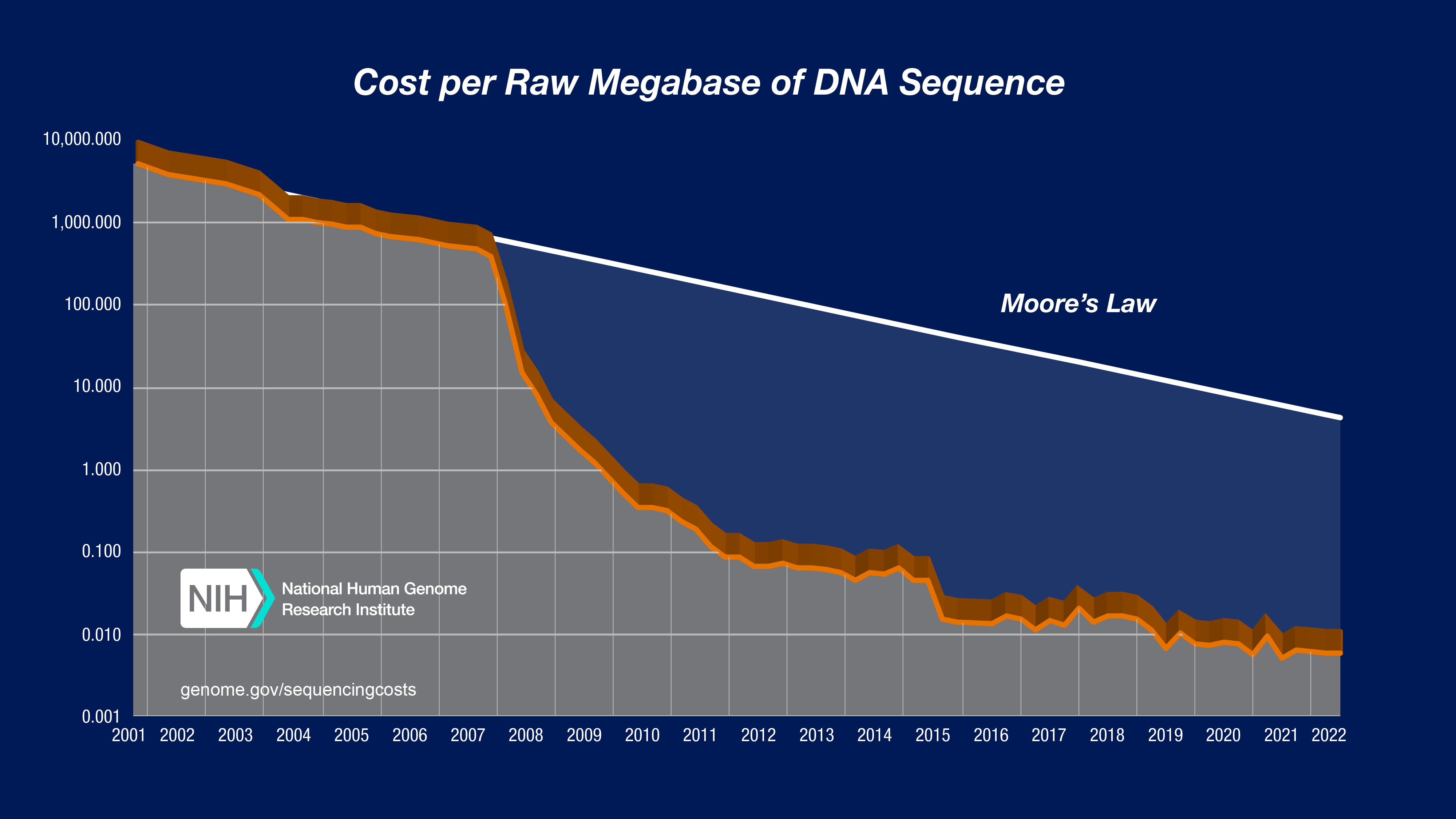
DNA sequencing to analyse genetic variation was introduced into the literature in 1983 by Kreitman (1983), based on a technology that was developed in the 1970’s by the British chemist Frederik Sanger.1
1 F. Sanger won two times the Nobel Prize for Chemistry, for developing a method for sequencing proteins and then for DNA sequencing. See the Wikipedia entry.
Currently, there are three major platforms available, all developed and marketed by commercial companies:
- Short read sequencing, i.e., reads from 100 to 300 bp in length (e.g., Illumina; http://www.illumina.com)
- Long read single molecule sequencing (long read sequencing) (e.g., PacBio; http://www.pacbio.com)
- Oxford Nanopore single molecule nanopore sequencing (long read sequencing) (https://nanoporetech.com/)
Short read sequencing
The Illumina technology is currently the leading technology in terms of volume and cost per base pair sequenced.
The technology is called sequencing-by-synthesis because new DNA molecules are synthesized during sequencing and the type of nucleotide that is incorporated in the newly synthesized DNA molecule is detected during the sequencing process (Figure 4)
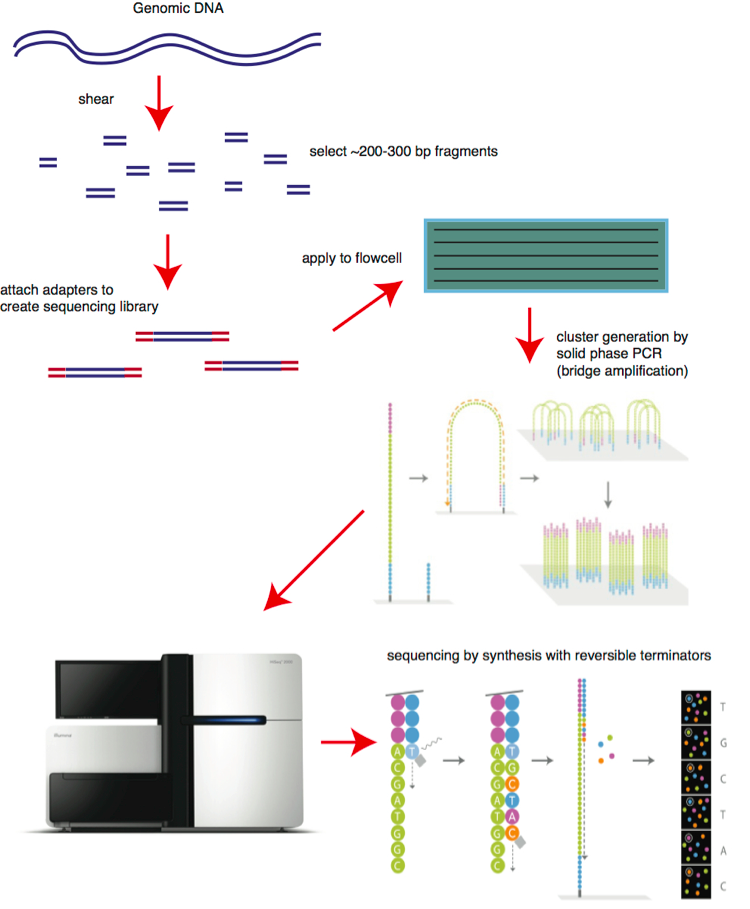
Although the fluorescent imaging system used in Illumina sequencers is not sensitive enough to detect the signal from a single template molecule, the major innovation of the Illumina method is the amplification of template molecules on a solid surface. The DNA sample is prepared into a sequencing library by the fragmentation into pieces each around 200 bases long. Custom adapters are added to each end and the library is flowed across a solid surface (the flow cell) and the template fragments bind to this surface. Following this, a solid phase bridge amplification PCR process (cluster generation) creates approximately one million copies of each template in tight physical clusters on the flowcell surface. Illumina has improved its image analysis technology dramatically which allows for higher cluster density on the surface of the flowcell.
A key characteristic of the Illumina technology is that reads are relatively short (up to 250 base pairs), which has some disadvantages, especially in the sequencing of repetitive regions of the genome.
The whole process can now be streamlined, and carried out on a large scale as shown in Figure 5.
Long read sequencing technologies
A more recent progress in sequencing technologies is the ability to sequence much larger pieces of DNA (kilobases to megabases long) in one piece. There are currently two major technologies that essentially operate by the same principle: A long DNA molecule is shuttled through a molecular (protein) pore and a state change at the pore caused by the different nucleotides is read out and converted into a basecall of a signal.
The key advantages of the new technologies are longer reads, which greatly facilitates the assembly of genomes, and a portability (to agricultural fields, for example) of the technology. Disadvantages in comparison to short read sequencing are a higher price, lower capacity and higher error rate.
However the technology is evolving rapidly, and substantial improvements can be expected in short time.
Check out the short introductory videos of the technologies:
- PacBio: https://www.youtube.com/watch?v=_lD8JyAbwEo
- Oxford Nanopore: https://www.youtube.com/watch?v=RcP85JHLmnI and a bit longer and with explanations: https://www.youtube.com/watch?v=qzusVw4Dp8w
Resequencing of populations
The rapid evolution of sequencing technologies and the low prices enable the whole genome sequencing of dozencs and hundreds of plant genotypes (or genebank accessions, or old landraces). This involves the sequencing of dozens and hundreds of individuals (genotypes) that have genomes of millions to billions of nucleotides each.
For example genome sizes of major crops are
- Rice: 500 Megabases (Mb)
- Maize 3000 Megabases (Mb)
Due to the huge amounts of data produced, the analyses are done mostly automatically with specialized computer programs.

Resequencing of whole genomes with this technology is now state of the art and carried out for numerous crop species.
Recent examples of such projects are the resequencing 121 wild and domesticated genotypes of the minor crop amaranth to investigate the process of domestication (Stetter et al., 2020) or a resequencing of 683 common bean genotypes to identify yield components (Wu et al., 2020).


Bioinformatics for sequence analysis
Short summary: Genotyping versus sequencing
What are the advantages and disadvantages of the two methods for studying genetic variation?
Genotyping with SNP arrays - Advantage: Only defined markers are used - Disadvantage: Ascertainment bias - The SNPs markers are selected from a small panel of genetic material and are then applied to a large panel of genotypes that may originate from different populations. Both the size and the composition of the SNP ascertainment panel has a strong effect on subsequent analyses.
DNA sequencing - Advantage: Complete genetic variation investigated - Disadvantage: Missing data, sequence assembly
To summarize, SNP genotyping is still very popular and important for plant breeding, i.e., the analysis of breeding populations in marker-assisted selection or QTL mapping of segregating populations. In contrast, DNA sequencing is now the method of choice for the analysis of natural populations, genebank material and complex breeding populations where the accurate determination of genetic relatedness and allele frequencies is important.
Bioinformatic analysis of sequencing data
After producing millions of short reads or long reads, the data need to be assembled into longer contiguous sequences reflecting the chromosome and assembled to identify genetic variants (Figure 9)
This is achieved with a set of bioinformatics tools that are used sequentially in sequencing workflows or bioinformatics pipelines, which are highly integrated with respect to input and output control (in particular, compatible file formats) and flow of analyses.
These pipelines have the following characteristics:
- The are usually run on Linux or Unix workstations or high performance computers (HPC)
- Specific pipeline software controls the order of programs and the integration of the output and intput of the various programs (Leipzig, 2016)
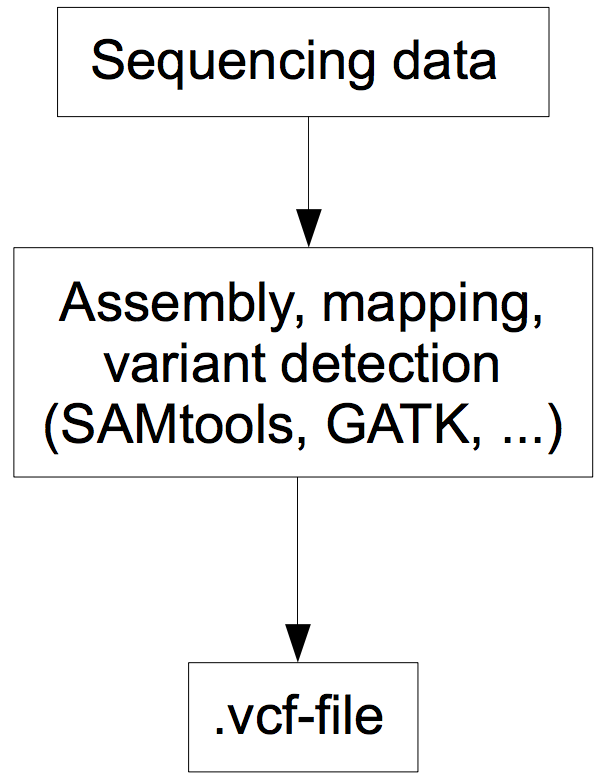
- The result of such a workflow is a file that contains the position of each sequenced nucleotide relative to a reference sequence. If multiple individuals are sequenced, the genetic variants are shown as well.
Figure 10 shows a section of a standard pipeline for sequencing, which illustrates that different file formats (e.g., BAM files) and multiple, independent programs (e.g., Picard) are used.
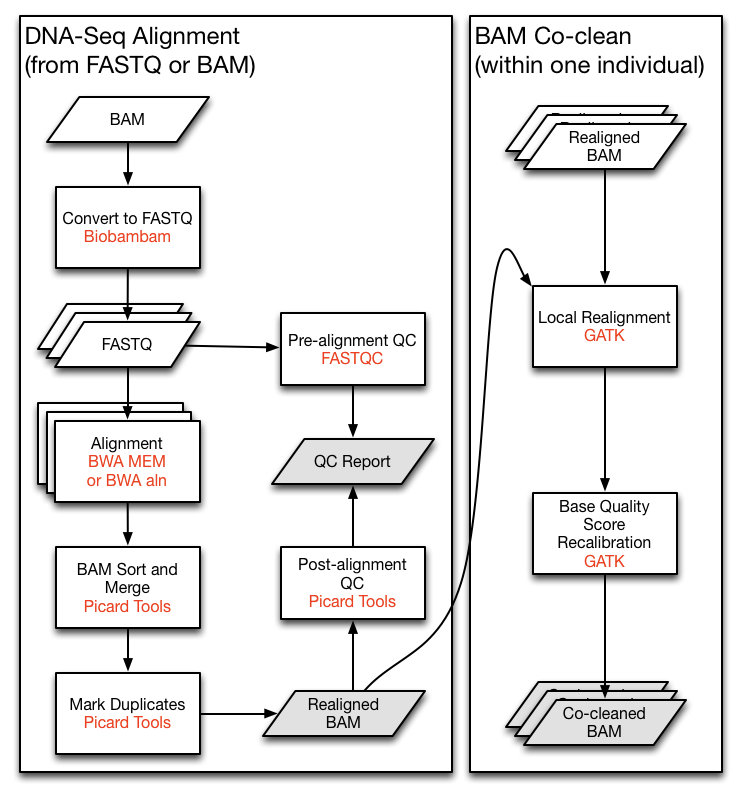
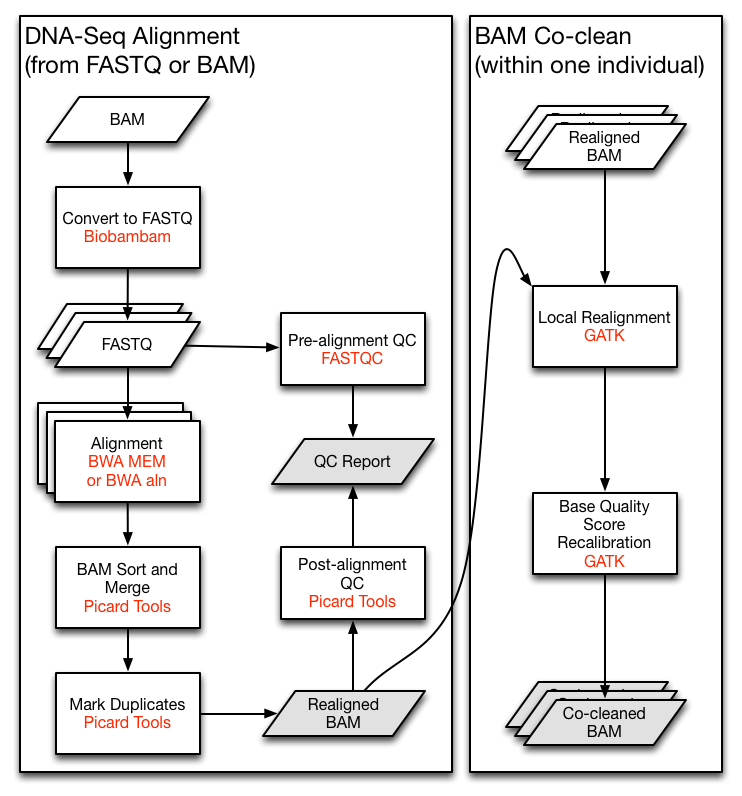{kind=link}
Variant Call Format (vcf)
The variant call format (vcf) is currently the most widely used format for storing sequencing data. It is essentially a text file format for representing variations like SNPs, indels or larger structural variants The current versionn is VCF format v4.2 and its format is described at https://samtools.github.io/hts-specs/VCFv4.2.pdf.
In the following, the VCF file format will be briefly described.
- VCF files consist of a header and a body.
- The header contains meta information about the file, which is indicated by
##.
The following types of meta information is included:
- File format, mandatory:
##fileformat=VCFv4.1 - Additional information:
##samtoolsVersion=0.1.18 (r982:295)
INFO lines with additional information:
##INFO <ID=DP,Number=1,Type=Integer,Description "Raw read depth">FORMAT lines:
##FORMAT <ID=GT,Number=1,Type=String,Description "Genotype">FILTER lines:
##FILTER <ID=q10,Description "Quality below 10" >Example of meta information:
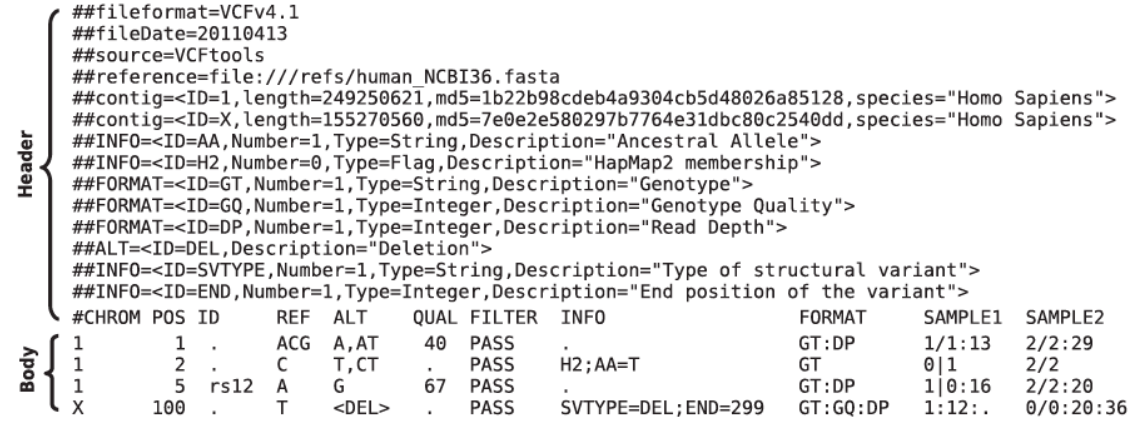
The header line has mandatory columns of 8 fixed fields:
CHROM- chromosome id or number POS - reference positionID- unique identifier(s)REF- reference base(s)ALT- alternative base(s)QUAL- phred-scaled quality score for ALTFILTER- filters passed or not passedINFO- additional information, specified in meta-information
An optional column is:
FORMAT- information about genotype, read depth, etc.
Figure 12 show how the different types of polymorphisms are represented:

Limitations of the VCF format
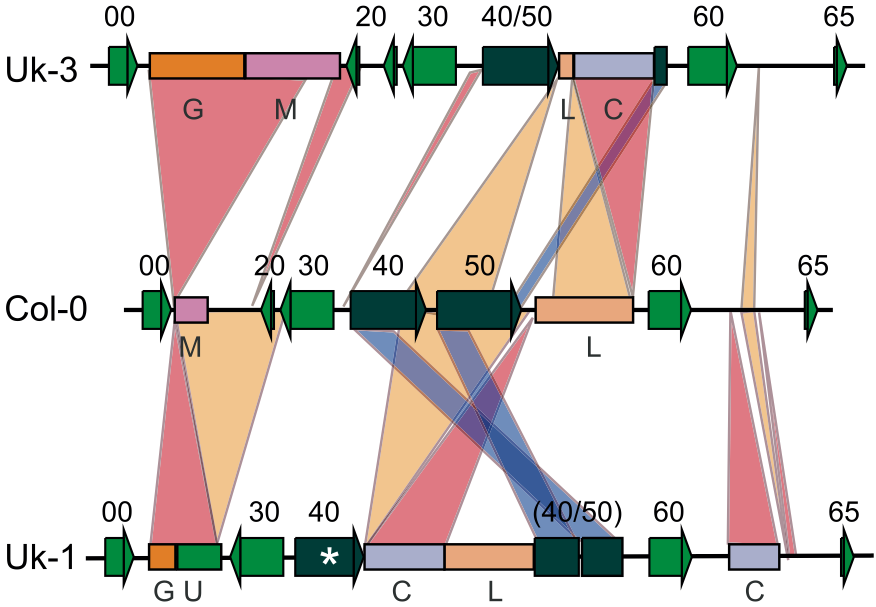
The VCF format is based on mapping short or long reads against a high-quality reference genome, which is used to determine the position of a polymorphism. Due to the repetitive nature of plant genomes, other, more complex polymorphisms and mutations arise. They include so-called structural variants, which can be nested (i.e., occur within each other). In such cases, a simple landmark in the genome, i.e., a position of a polymorphism at a given position in a chromosome can not be identified because the regions can not be aligned, i.e., compared with each other and even have now shared evolutionary history (they are not homologous) because gene duplications or gene loss of whole or partial genes may have occured. This process can either reflect neutral evolution or natural selection. In the latter case, it may be advantageous to be highly polymorphic at a locus, a phenomenon that is very frequently observed at genes involved in disease resistance. Complex structural rearrangements in multiple individuals of the model plant Arabidopsis thaliana in a region of the genome that encodes genes that recognize bacterial and fungal pathogens (Figure 13)
Working with VCF files
Although vcf-formatted files are human-readable, they are usually very large (Megabases to Gigabases) so that they are only processed by computer programs. There are multiple software tools available that allow to work efficiently with vcf-formatted files.
Examples are:
- vcftools: Written in Perl, not so fast (http://vcftools.github.io)
- bcftools: Written in C very fast, fewer functions (https://samtools.github.io/bcftools/)
These are commandline tools, but multiple libraries in the R, Python and Julia languages are available to parse, manipulate and analyse vcf-formatted files.
Summary
- Genetic variation is abundant in plant genomes and useful for the characterization of the material
- There are different types of variants: Single nucleotide variants (SNPs), Indels and structural variants (SVs)
- Mutations cause new variation; Polymorphisms are mutations that segregate in a population; and markers are variants that can be detected with a molecular assay.
- The most important methods for the characterization of genetic variation are genotyping assays and genome sequencing.
- Due to the decreasing prizes of sequencing, whole genome resequencing is become the method of choice for the analysis of genome variation in crop plants.
- The large amounts of data resulting from sequencing projects are analysed with bioinformatics pipelines.
- These pipelines usually involve a large number of programs.
- The variant call format (VCF) is a machine readable file format to store genetic variation data and allows to store different types of polymorphisms.
Key concepts
Further reading
- Ekblom and Wolf (2014) - A very good starter on sequencing technologies for studying genetic diversity
On the importance of nanopore sequencing for plant genomics:
Study questions
- Why do different types of genetic variants exist in plant genomes?
- What are the advantages and disadvantages of SNP genotyping?
- What are the advantages and disadvantages of DNA sequencing?
- What are the differences and respective advantages of short read and long read sequencing?
- What are the relative advantages of short and long reads for the analysis of genome variation?
- What are the challenges of the VCF file format?
- What is the challenge for comparing highly polymorphic loci with many SNPs or InDel polymorphisms that can not be aligned well?
- What information is included in a typical Variant Call Format (VCF) file? Briefly describe the structure of a VCF file, including at least five key columns that are always present.
Problems
Choosing a genotyping technology
You have been tasked with studying a crop species for which no genetic information is available. Your objective is to select the most suitable genotyping technology for this scenario. Should you choose SNP arrays or whole-genome sequencing, and why?
Different sequencing approaches
Sequencing has become more and more important and is part of many scientific studies. In this exercise you will investigate some studies that used different sequencing approaches in order to familiarise yourself with it.
In the following, there is a list of publications:
Find out which genetic material the authors sequenced, which methodology they applied and what the purpose of the sequencing was.
Specifically, you should try to find out the following:
- What was the purpose of sequencing in this study?
- What sequencing approach, methodology, and technology was applied?
- Where was the sequencing conducted?
- Details about the experimental set up:
- how many samples?
- which read length and type?
- how many reads, or how much sequencing output was obtained?
- what was sequencing depth (/i.e./ coverage)?
- Which softwares did the authors use for data processing and was a reference genome available?
- Where has the data been deposited?
Determining the total output for a sequencing project
For some new project, your research group wants to sequence 25 genotypes of sugar beet (Beta vulgaris). After some discussion, it was decided to use the Illumina short read technology with 150 bp read length and paired end reads, and to sequence each sample at 10x coverage (i.e. sequencing depth).
- Calculate the required total output in Gigabases (Gb). The genome size of sugar beet is about 740 Mb.
- Why or for what reason could it be important to calculate the required total output in Gb? Think about practical aspects.